PLANT ANATOMY LAB I
THE COMPOUND AND DISSECTING MICROSCOPES
Note:
Links that are italicized are to Animated GIF89A files.
You should wait for these to execute when you are viewing them. (STEP by
STEP) Links break these animations down to frame by frame viewing for your
educational pleasure.
Obiectives
The most important tools in your study of
plant anatomy will be the compound and the dissecting microscopes. Although
for most of you this exercise will be review, you should take the time
to read about the care and use of these microscopes before beginning the
other exercises. You will obtain much more from all the exercises that
follow if you first familiarize yourself with the "tools of the trade".
Indeed, this is the only way to proceed.
General Description
The conventional compound
microscope used in biology consists of a light source, a condenser
lens with an adjustable aperture, specimen slide for trans-illumination
specimens, an objective lens and an ocular lens plus appropriate mechanical
fixtures for focusing and mounting specimens. This
exercise purposesly uses an unfamiliear microscope to test your understanding
of the basic components and functions of all microscopes. Once you
understand the functional reason of each microscope component, it won't
matter who manufactured the microscope, you will be able to evaluate any
microscope by your criteria! So shoddy microscope sales people won't
be able to sell you shoddy microscopes!
The illumination
system is designed to fill both the condenser and objective apertures,
if Kohler illumination is maintained. Objectives are designed to produce
a real image of the specimen of a quality which approaches the theoretical
limit imposed by the wave-nature of light. Oculars are designed to produce
(a) a virtual image which may be observed visually or (b) a real image
which can be projected onto a screen or photographic plate. Efficient use
of the microscope and interpretation of images required consideration of
the following factors:
-
1. Magnification
-
2. Resolving power
-
3. Depth of field
-
4. Contrast
-
5. Practical errors in lens design and manufacture
Magnification,
when the image is viewed directly, is the ratio of the tangent of the angle
subtended at the eye by the virtual image compared to the tangent of the
angle subtended at the eye by the object when viewed at 25 cm from the
eye. This value is approximately the product of the nominal objective and
ocular magnifications; e.g. 40X objective with 10X ocular = 400 total magnification.
In the case of a projected image, magnification is simply the ratio of
the linear dimensions of the image to the corresponding dimensions of the
object. If the image is projected a distance of 25 cm from the ocular the
magnification will be, as above, the product of the nominal magnifications
of the objective and ocular. At other projection distances this value
is multiplied by the projection distance in cm divided by 25. Critical
use demands the selection of a magnification such that the finest details
of the image (determined by the resolving power of the microscope) can
be resolved by the retina or photographic plate. Magnification which exceeds
this is known as "empty magnification" since it reveals no additional detail.
Objectives are usually designed so that persons with normal vision can
see all that can be seen by using a 10X or a 15X ocular.
Resolving power
(STEP By STEP) is defined as the
least distance between points in the object (d) that can be distinguished
in the image. For a perfect lens with a circular aperture, the resolving
power is given (according to Rayleigh' s criterion for "distinguishability")
by:
d = 0.6 l
/ (n* sin(q))
where d is resolvable distance given in
the same units of length as l
; l is the
wavelength of light; n is the index of refraction of the material in which
the specimen is embedded and q
is the half angle of the cone of light accepted by the lens when this angle
is measured at the specimen.
This expression for resolving power applies
to any kind of lens (telescope, camera, microscope, eye, etc.) but it assumes
certain conditions of illumination that are not automatically met when
a lens is used as a microscope objective. To satisfy these conditions the
cone of light illuminating the specimen must equal the cone of light accepted
by the lens.
For a lens used at a single set of object
and image distances (as in a microscope) the quantity (n sin q)
is a constant known as the numerical aperture (N.A.) of the lens., Most
microscope objectives have the values of the N.A. engraved on the lens
barrel along with the focal length and/or the magnification of the lens.
The resolving power defines a limiting accuracy with which lateral dimensions
of a specimen can be measured.
Depth of field, D, is the thickness
of the specimen, measured along the optical axis, which is in sharp focus
for any one set of conditions. It is given approximately by the formula
D = 2r*cot(q).
2r is the diameter of the Airy disc, see below, and
q is as defined above. Note that
for practical purposes d of the Abbe equation is equal to 2r of the
depth of field equation. As the aperture angle q,
increases, the depth of field decreases to a limiting value of about 0.5
microns with the oil immersion objective .
Depth can be measured with the compound
light microscope. Knowing the depth of field associated with particular
lenses, which is a small fraction of the thickness of a typical biological
specimen, makes possible the "opt ical sectioning" of specimens.
CAUTION --
Beginners often restrict the aperture of the microscope by closing the
condenser of the microscope because of a subjective impression that they
can see more. They also tend to increase the apparent depth of field by
forcing the eye to accommodate for failure to use the fine focusing adjustment.
These "techniques" do not enhance available information and often result
in eye strain and headaches.
Contrast depends on the signal to
noise ratio and upon the physiology of vision. In this case the signal
is the image of interest and the noise is background of light associated
with out-of-focus images, light scattered by dirt on optical surfaces,
etc. Detectable contrast depends upon the ability of the eye (or of any
other measuring devise) to make fine distinction in intensity and color
on resolvable areas of the image. In general the greater the noise the
greater the signal intensity required to produce a detectable image. Texts
in human physiology should be consulted on this point. Nearly all histological
and cytological techniques have been developed to compensate for the fact
that living cells in visible light display very little contrast
.
Practical errors. While the best
of the modern microscope lenses approximate very closely the theoretical
limits of a "perfect lens," most introduce compromises in design and defects
in manufacture. By convention the resulting errors are classified as follows:
-
1. Airy disc, circle of confusion.
It is an inherent property of waves that the image of a point cannot itself
be a point, but must be a small circle, the Airy disc. Thus a point cannot
be distinguished from a small circle which is called the circle of confusion.
Any two points which fall within the radius (not the diameter) of the circle
of confusion cannot be resolved as separate points. The Airy disc is the
circle of confusion of the image. For the normal eye at normal viewing
distance from a print or a photograph, the circle of confusion is between
0.1 and 0.2 mm and any print with circles a confusion less than this will
appear sharp.
-
2. Chromatic aberration. This
aberration is the inability of the lens to bring rays of different wavelength
to the same focus. Modern Achromatic lenses are designed to correct this
aberration for blue and yellow wavelengths of visible light. The more expensive
Apochromatic lenses are designed to correct this aberration for all visible
light wavelengths.
-
3. Spherical aberration. This
aberration arises from the inability of a lens to bring those rays close
to the axis to the same focus as those more distant from it, and effectively
limits numerical apertures to a very small angle. Aplanatic lenses are
designed to correct this aberration, thus making higher numerical apertures
possible.
-
4. Astigmatism. This aberration
arises from imperfection in the radial symmetry of the lens such that the
image is distorted along one diameter of the lens. Most good lenses have
sufficient quality that this aberration is minimal.
The above errors are interrelated so that
typically when one defect is present other types of defects will also be
found. The effect of these errors upon the quality of an image must be
understood before the results of observations and measurements can be interpreted.
The best lens (and most expensive) one can buy is an Aplanatic Apochromatic
lens. Most good quality research microscopes are equipped with such lenses.
Specif ic Description
Two types of light microscopes (as distinct
from electron microscopes) will be employed during our laboratory sessions.
Most often we' ll use standard binocular compound microscopes; those available
in this laboratory have a built-in light source, and provide magnifications
of 100, 400, or 1000 diameters, depending upon which of the three objective
lenses of the revolving nosepiece is in place under the main tube. The
ocular (eyepiece) magnifies 10X, the objective lenses 10, 40, and 100X,
respectively, hence the overall magnification is the product of the magnification
provided by the ocular and that provided by the objective lens.
The 10X magnifying objective lens should
be used first, and only then if additional magnification is required should
one shift to the 40X (high-dry) objective lens. These lenses are essentially
parfocal, which means that only little focusing is necessary in shifting
from one objective lens to another. The 100X objective is an oil immersion
lens , which means that a clear image can be obtained only if a special
type of oil (immersion oil) is used to bridge the space between the bottom
element of this lens and the cover glass and between the top of the condenser
lens and the bottom of the specimen slide. If you have not used one previously,
please ask the laboratory instructor to show you how. After using
the oil immersion lens it is essential that both the condenser and
objective lenses be thoroughly cleaned!
CAUTIONARY NOTES:
-
1. When you return the microscope
to the cabinet be certain that either of the two lower-magnification objective
lenses is in place, and that these are at least a centimeter above the
stage.
-
2. Unless the surfaces of the cover
glass, the bottom lens of the objective, and the condenser lens are absolutely
clean the image will be blurred; therefore it is a good plan to wipe these
surfaces with lens paper before (and after) using the microscope.
-
3. The light microscopes you will
be using are equipped with phase optics. Phase contrast microscopy
is a useful technique for enhancing the contrast of unstained biological
material. These optics utilize a special phase ring, inserted below the
condenser, in combination with an objective containing a phase annulus.
(This 40X objective has a red ring about its barrel and should not be used
for bright field microscopy.) Since you will be using bright field
optics almost exclusively in this laboratory it is necessary to learn how
to properly remove the phase ring from the condenser. This is accomplished
by grasping the phase ring insert at the bottom on the condenser with thumb
and forefinger while holding the condenser firmly with the other hand.
Gently pull the phase ring insert out of the condenser making sure that
the condenser stays intact.
REMEMBER : THESE
OPTICS ARE DELICATE AND EXPENSIVE. CAUTION SHOULD BE TAKEN NEVER TO DROP
ANY OF THE MICROSCOPE COMPONENTS OR TO PLACE ANY LENS SYSTEM IN SUCH A
WAY THAT ITS SURFACE CAN BE SCRATCHED.
To insert the phase ring insert after you
have finished the current exercise reverse the above procedure. BE SURE
TO REPLACE THE PHASE RING INSERT BEFORE RETURNING THE MICROSCOPE TO THE
STORAGE CABINET.
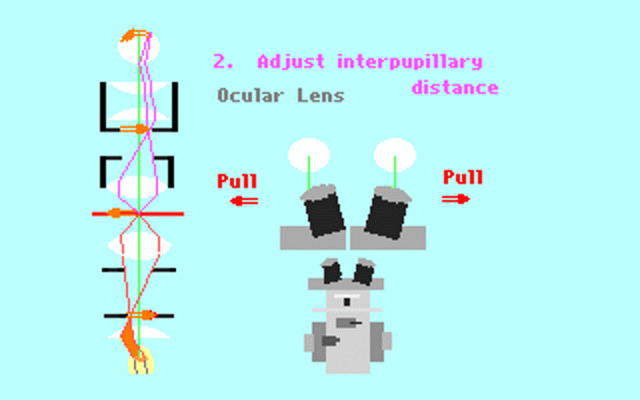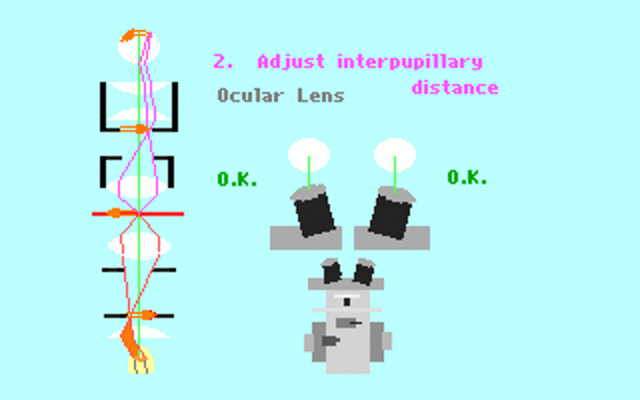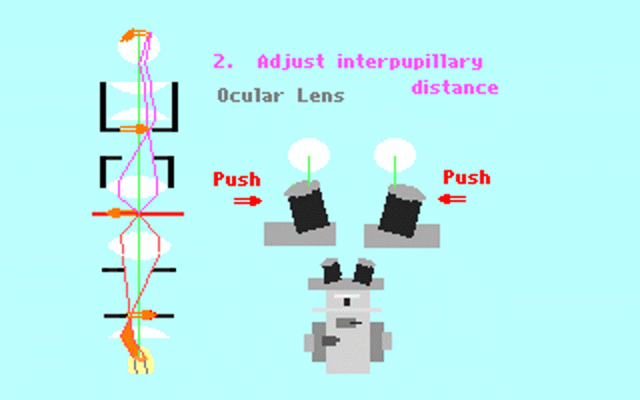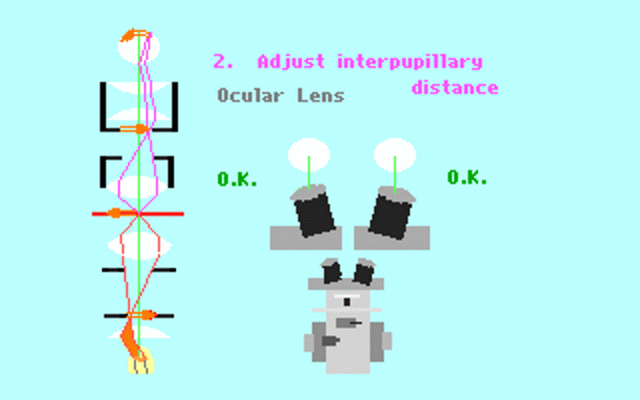
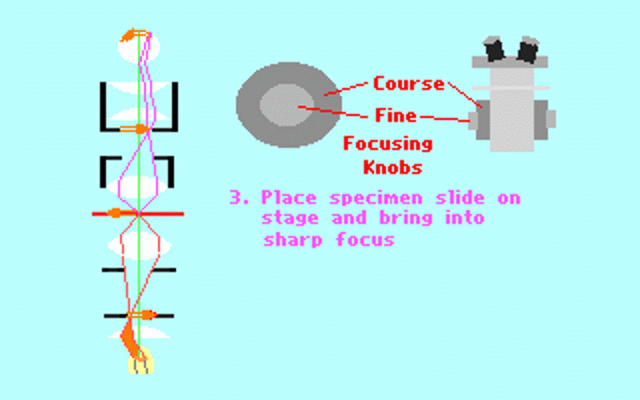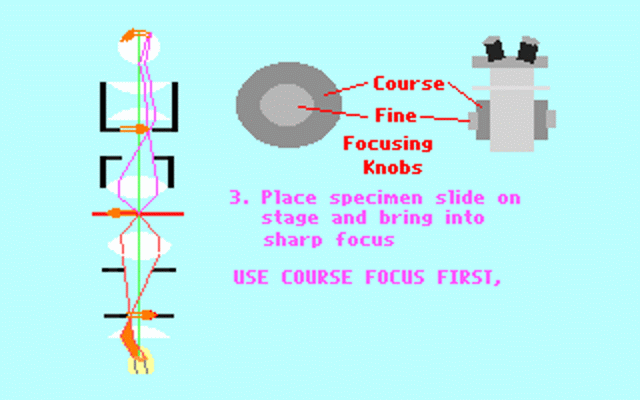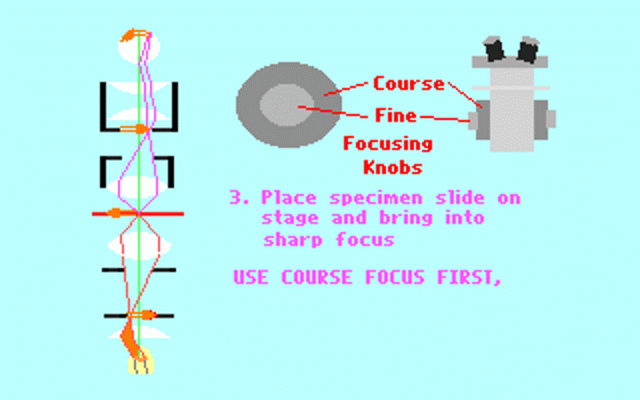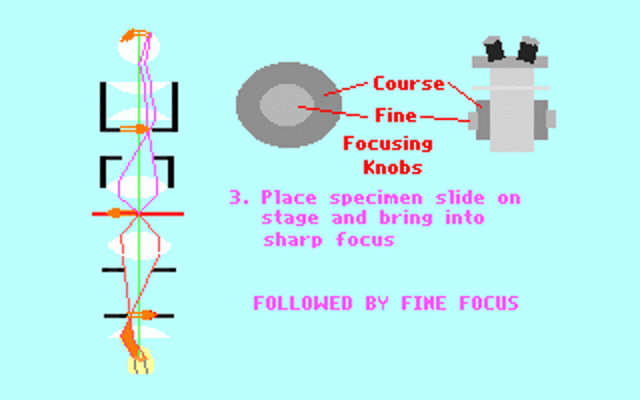
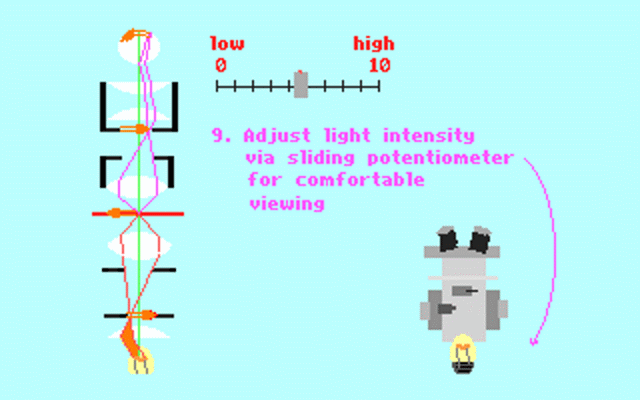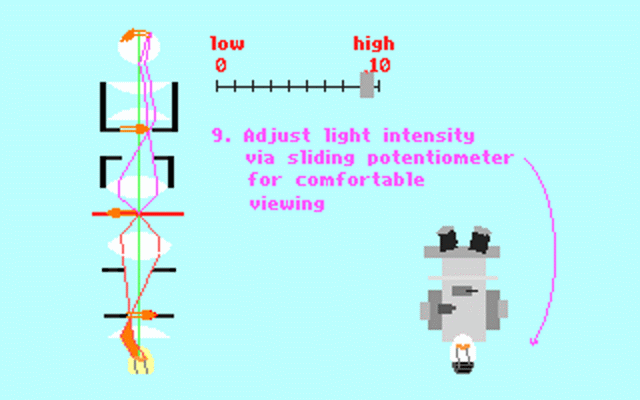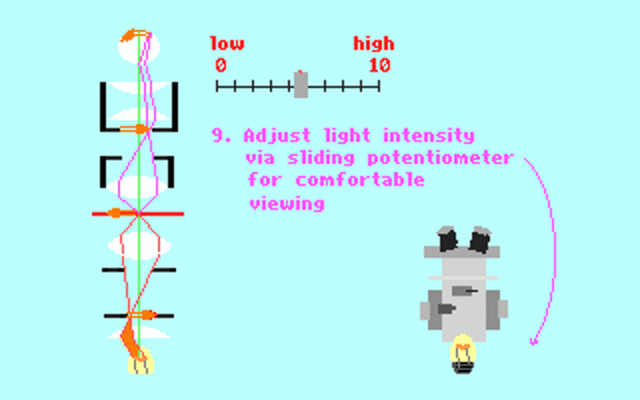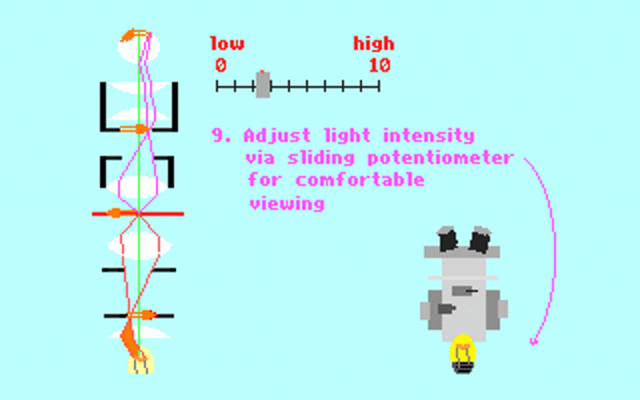
Kohler illumination procedure:
To obtain the optimum resolution of the light
microscope precise control of the light path should be obtained. The accepted
method for this is termed Kohler illumination after August Kohler who developed
the theory and procedure in 1894. This procedure should be preliminary
to each exercise session of this laboratory. Click on each link to
obtain additional help.
NOTE : When the
objective lens is changed it is necessary to adjust the condenser diaphragm
to 80% illumination as in steps 10-13.
BE SURE TO FOLLOW
EARNST ABBE'S ADVICE TO AVOID EYE STRAIN AND SUBSEQUENT HEADACHES!
The other type of microscope that we' ll
use occasionally is the dissecting binocular or stereoscopic microscope.
Illumination units which can be used either above or below the stage are
available. These microscopes (or at least some of them) have zoom facilities,
which provide a continuous range of magnification. This type of microscope
is used at relatively low magnification for dissection, and to show the
three-dimensional nature of what is observed.
Exercises:
These should be completed using Netscape Composer
to modify this exercise sheet.
1. Obtain a compound
light microscope and review its component parts.
2. Work through the Kohler illumination
procedure until you can establish this in less than 1 minute.
3. Use the stage micrometer to calibrate
the mechanical stage micrometer for your microscope. The stage micrometer is a vernier scale. Verify your calibration
at all magnifications. The well-known distance formula:
distance
= sqrt [ (x1 - x2)2 +(y1 -
y2)2 ]
from analytic geometry will be useful
to keep in mind for scale calculations in future labs.
4. Calculate the resolution for the 10X,
40X and 100X objectives you' ll be using.
5. Calculate the depth of field of the
10X, 40X and 100X objectives you' ll be using. Learn how to estimate specimen
depth using the fine focus scale.
Materials
1. Olympus
Student Microscope Instruction Manuals
2. Fake
field diaphragms and lens paper
3. Stage
micrometer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
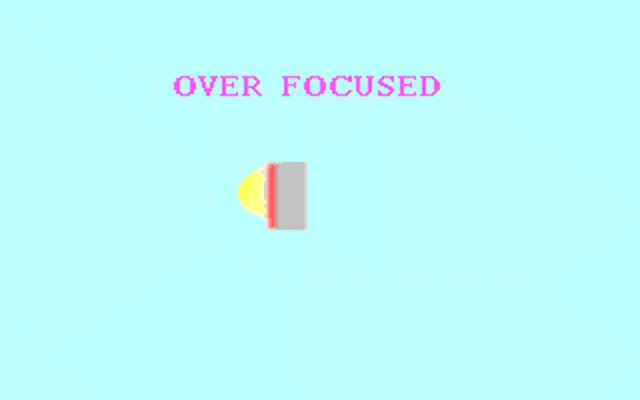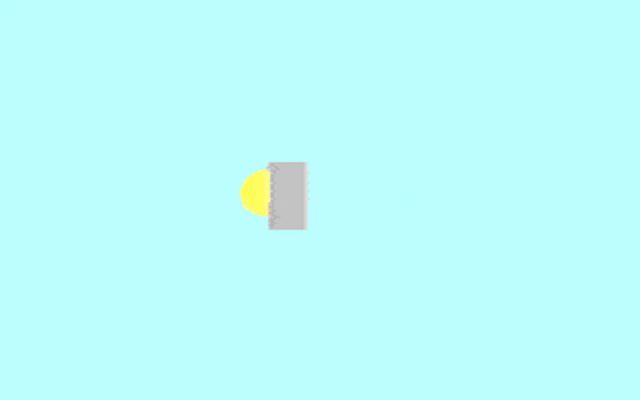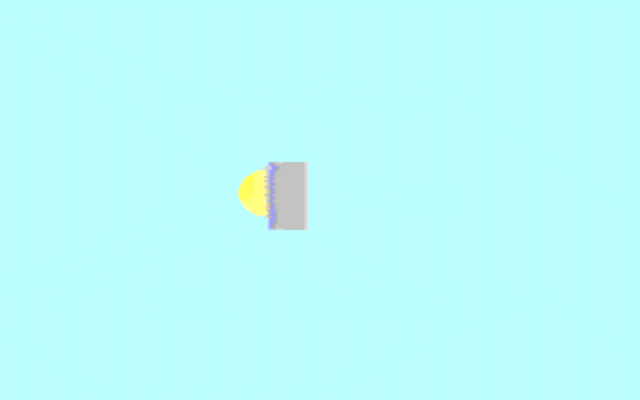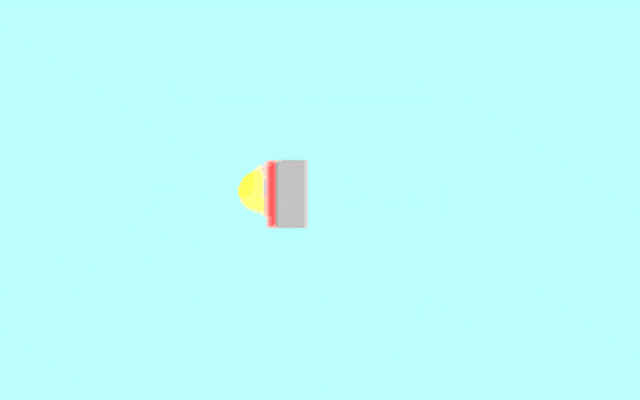{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}