
Direct Allelic Variation Scanning of the Yeast Genome
Elizabeth A. Winzeler,* Dan R. Richards, Andrew R. Conway, Alan L. Goldstein, Sue Kalman, Michael J. McCullough, John H. McCusker, David A. Stevens, Lissa Wodicka, David J. Lockhart, Ronald W. Davis
As more genomes are sequenced, the identification and characterization of the causes of heritable variation within a species will be increasingly important. It is demonstrated that allelic variation in any two isolates of a species can be scanned, mapped, and scored directly and efficiently without allele-specific polymerase chain reaction, without creating new strains or constructs, and without knowing the specific nature of the variation. A total of 3714 biallelic markers, spaced about every 3.5 kilobases, were identified by analyzing the patterns obtained when total genomic DNA from two different strains of yeast was hybridized to high-density oligonucleotide arrays. The markers were then used to simultaneously map a multidrug-resistance locus and four other loci with high resolution (11 to 64 kilobases).
E. A. Winzeler,
D. R. Richards, A. R. Conway, S. Kalman, R. W. Davis, Department of Biochemistry,
Stanford University School of Medicine, Stanford, CA 94305-5307, USA. A. L.
Goldstein and J. H. McCusker, Department of Microbiology, 3020, Duke University
Medical Center, Durham, NC 27710, USA. M. J. McCullough and D. A. Stevens, Department
of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA.
L. Wodicka and D. J. Lockhart, Affymetrix, 3380 Central Expressway, Santa Clara,
CA 95051, USA.
* To whom correspondence should be addressed. E-mail: winzeler@cmgm.stanford.edu
These authors contributed equally to the work.
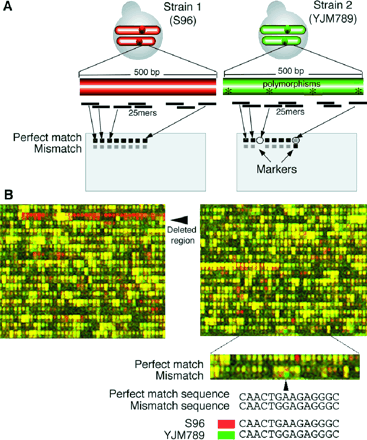
Figure 1. (A) Detecting allelic variation with high-density arrays. For nonduplicated
regions of the genome, a minimum of 20 25-base oligonucleotide probes was chosen
from yeast genomic sequence (S288c) for every annotated ORF in the yeast genome
(3). Probes (only from predicted coding regions) were generally arranged on
the array in order of their chromosome position. In addition to probes designed
to be perfectly complementary to regions of yeast coding sequence (PM), probes
containing a single base mismatch in the central position of the oligonucleotide
were also synthesized in a physically adjacent position. The mismatch probes
serve as background and nonspecific hybridization controls in other analyses
(3, 31). If probes complementary to YJM789 DNA fragments containing polymorphisms
(*) are found on the array, decreases in signal intensity at these probes relative
to the S96 signal may be observed when YJM789 DNA is hybridized to the array.
The amount of signal decrease will depend on several factors, such as initial
probe intensity and whether the probed fragment is completely absent in YJM789
or contains a small substitution. The location of the polymorphism within the
probe sequence will also affect the observed intensity decrease. (B) Comparative
genomic DNA hybridization patterns. Genomic DNA from two strains of S. cerevisiae,
YJM789 and S96, was fluorescently labeled and hybridized to two different arrays.
Scanned images of the arrays were collected, digitally colored red or green,
and then electronically superimposed. A portion of the composite image is shown.
Probes that hybridized to S96 DNA more efficiently than YJM789 DNA are red,
and probes that hybridize to both DNA samples with equal intensity are yellow.
A region that is completely deleted in YJM789 is indicated by an arrow. The
figure closeup shows a region in which one of the mismatch features is bright
green. Shotgun sequencing of YJM789 demonstrated that the actual sequence of
YJM789 was complementary to the sequence of the oligonucleotide in the mismatch
row and not to that in the perfect match row.
Text iGenetics by Peter J. Russell
This web site is provided for instruction in Botany and Zoology 342
by Kenneth G. Wilson,
Professor of Botany
Miami University
wilsonkg@muohio.edu